CIPHER-SKI Compiler Infrastructure for Persistent Homology Enabled Rewriting – Scaffold Kinetic Insight
Rewriting Chemistry as Code: The LLVM of Drug Discovery
CIPHER-SKI is not a generative model. It is the LLVM of drug discovery, a full compiler stack whose source language is chemistry, whose target language is biologically active, topologically rich, drug-like molecules, and whose optimization objective is “maximize interesting 3D structure + therapeutic utility.”
Just as a modern compiler takes source code, applies safe rewrite rules, runs intelligent optimization passes, and emits efficient machine code, CIPHER-SKI takes simple molecular fragments and intelligently rewrites them into optimized drug candidates using the exact same principles that power today’s most advanced programming compilers.
While energy-based modeling has gained traction in machine learning as a foundation for structured reasoning, most existing approaches remain confined to continuous latent spaces. We introduce a framework that treats molecular design as a compilation process, analogous to how LLVM compiles high-level programs into optimized machine code. In this model, graph rewrite rules serve as the intermediate representation, while energy functions defined over these rewrites, guided by the partition function and Boltzmann sampling, act as the optimizer. By constructing hybrid controllers that combine thermodynamic priors with learned policies, our system generates molecules through a sequence of principled, energy-directed transformations rather than purely statistical pattern matching.
The Problem
Most AI drug discovery tools remain stuck in incremental generation mode. They remix known chemistry, produce relatively flat, aromatic-rich libraries, and deliver molecules that often feel more like statistical outputs than the thoughtful, intuitive designs of experienced medicinal chemists.
The result is predictable: weak three-dimensional character, poor developability, high late-stage failure rates, and significant resources spent on compounds that ultimately struggle to advance. The industry doesn’t need better generators. It needs a new compiler paradigm.
Our Solution: The Molecular Compiler
CIPHER-SKI treats every molecule as an executable computer program that can be safely rewritten, intelligently optimized, and deeply evaluated for structural beauty and real-world performance. It operates through a clean two-phase compiler architecture that mirrors how successful medicinal chemistry teams actually work:
Phase 1 – Smart Scaffold Hopping Creatively redesigns the molecular core using chemically valid graph-rewrite rules and advanced 3D topological analysis via persistent homology. Only candidates with genuine three-dimensional richness long-lived cavities, spiro systems, and bridged rings, survive to the next stage.
Phase 2 – Branch Optimization Adds realistic, ADME-friendly side chains drawn from clinically validated fragment libraries (ring systems + NCEs), while intelligently balancing binding affinity, drug-likeness (QED), synthetic accessibility, Fsp³ (3D character), and overall developability.
The system learns from every discovery. It remembers which molecular cores perform best and gradually favors them, while still retaining the ability to make bold architectural jumps when needed.
How It Works: The Three Compiler Phases
At its core, CIPHER-SKI follows a classic compiler pipeline, enriched with domain-specific tools for chemistry:
- Front-End (Molecular Parsing & Validation) Parses fragment libraries into rich molecular graphs, performs chemical “type checking” (sanitize), and builds a high-dimensional state embedding via a Graph Neural Network (GNN).
- Middle-End (Intelligent Optimization) Applies safe graph-rewrite rules (the reaction templates) guided by a Deep Q-Network (DQN) policy. A persistent-homology scaffold ranker acts as a global semantic filter, aggressively favoring molecules with long-lived 3D cavities, bridged systems, and high Fsp³ – exactly the topological features that make leads interesting to medicinal chemists.
- Back-End (Emission & Retrosynthetic Lowering) Produces final, sanitized, drug-like molecules optimized across binding affinity, QED, synthetic accessibility, ADME properties, LogP, TPSA, molecular weight, and 3D character, ready for synthesis planning and biological execution.
This tightly integrated, physics-informed compiler goes far beyond statistical pattern matching. It actively compiles structurally novel, realistic chemical matter guided by real physical principles, topological aesthetics, and medicinal chemistry knowledge.
Why CIPHER-SKI Matters
- Not Generation — Compilation Treats molecules as executable programs that can be safely rewritten, intelligently optimized, and deeply evaluated on structural beauty.
- True Scaffold Hopping Generates entirely new chemical series with rich 3D topology rather than incremental analogs or flat aromatic libraries.
- Persistent Homology as Shape Typing Uses H1/H2 persistence as a first-class semantic signal to reward genuine 3D complexity; cavities, spiro systems, and bridged rings that medicinal chemists instinctively recognize as high-potential leads.
- Superior Lead Quality Produces molecules that simultaneously achieve strong predicted binding, high drug-likeness, synthetic accessibility, and the distinctive “spiro-rich, bridged, 3D” aesthetic prized by experienced drug hunters.
- Exceptional Efficiency Curriculum-trained reinforcement learning enables rapid, stable exploration of novel chemical space with limited data.
The Deep Insight: A molecule is a computing system
In functional programming compilers such as GHC (Haskell) or LLVM, programs are represented as graphs and transformed through libraries of safe rewrite rules to produce faster, cleaner code while preserving validity. CIPHER-SKI does exactly the same thing, but applied to molecules instead of code:
- Atoms and bonds become the graph
- Medicinal chemistry reaction templates become the rewrite rules
- Persistent homology becomes the higher-order semantic analyzer (shape typing)
- The Deep Q-Network becomes the learned super-optimizer
This is why CIPHER-SKI molecules feel different. They are not sampled, they are compiled. CIPHER-SKI represents a paradigm shift: drug discovery as compilation, not generation. By internalizing both the rules of chemistry and the aesthetics of interesting molecular topology, it delivers leads that look and behave like they came from a world-class medicinal chemistry team – because, in a very real sense, they did.
Proven Performance
Early validation on the notoriously difficult Switch-II pocket of KRAS G12C (PDB 6OIM) has already shown outstanding results. CIPHER-SKI generated multiple novel scaffolds with predicted binding affinities reaching -11.1 kcal/mol while maintaining favorable drug-like properties (good QED, LogP, and synthetic accessibility).
These results highlight the platform’s ability to tackle historically “undruggable” targets where conventional and purely statistical AI methods have struggled.
CIPHER-SKI: AI-Driven Molecular Discovery
Generated Molecules Successfully Docked into PDB 1EIP
CIPHER-SKI combines a hybrid AI architecture powered by Graph Neural Networks and Deep Reinforcement Learning (DQN) to perform true scaffold hopping.
The system is guided by a sophisticated multi-objective scoring function that integrates predicted binding affinity from molecular docking, drug-likeness (QED), synthetic accessibility (SA Score), and other key physicochemical properties.
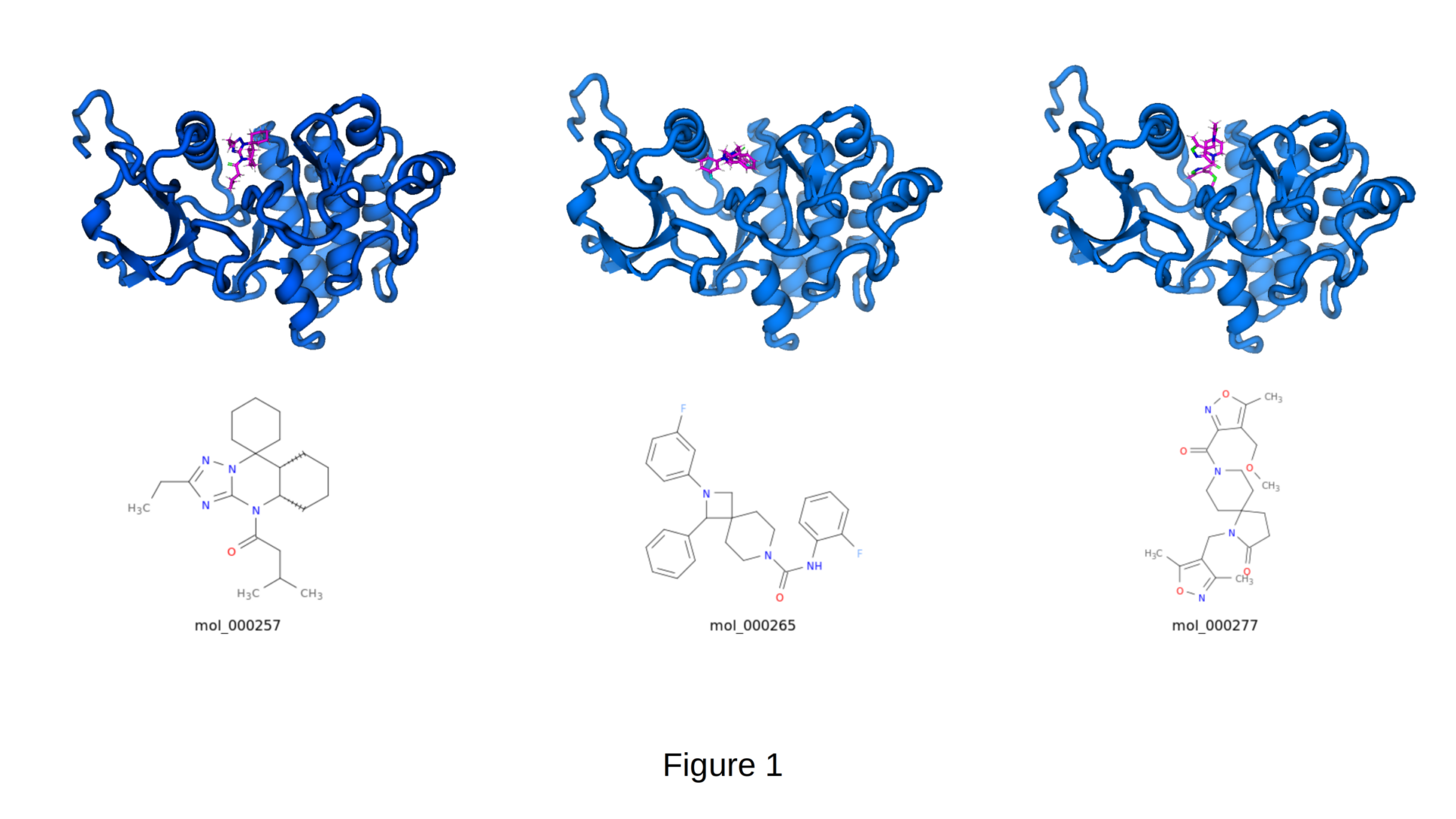
The three molecules shown in Figure 1 were autonomously discovered and optimized by CIPHER-SKI. They demonstrate high shape complementarity and favorable interactions within the binding pocket of PDB 1EIP.
Highlighted Molecules:
- mol_000257: Compact spirocyclic scaffold with constrained conformation and excellent steric fit.
- mol_000265: Fluorinated diaryl derivative exploiting hydrophobic regions of the binding site.
- mol_000277: Complex heterocyclic spiro-system featuring multiple hydrogen-bond acceptors for enhanced target engagement.
Key Capabilities Demonstrated:
- Novel scaffold generation beyond traditional virtual screening libraries
- Physics-guided optimization (docking + molecular dynamics minimization)
- Strong balance between binding affinity, drug-likeness, and synthetic accessibility
- Fully explainable chemical transformations via explicit pattern-based rewriting
Results on the 4XV2 Target (BRAF V600E Mutant Kinase)
The PDB structure 4XV2 represents the crystal structure of the human BRAF kinase carrying the V600E oncogenic mutation, the most common and clinically aggressive driver mutation in melanoma and several other cancers. This mutant form constitutively activates the MAPK signaling pathway, promoting uncontrolled cell proliferation and tumor progression. The binding pocket of BRAF V600E is notoriously challenging: it is relatively shallow, highly flexible, and features a complex mix of hydrophobic, polar, and hinge-region interactions that make traditional scaffold-based design difficult. Many existing BRAF inhibitors suffer from resistance mutations or limited selectivity precisely because they rely on a narrow set of chemotypes.
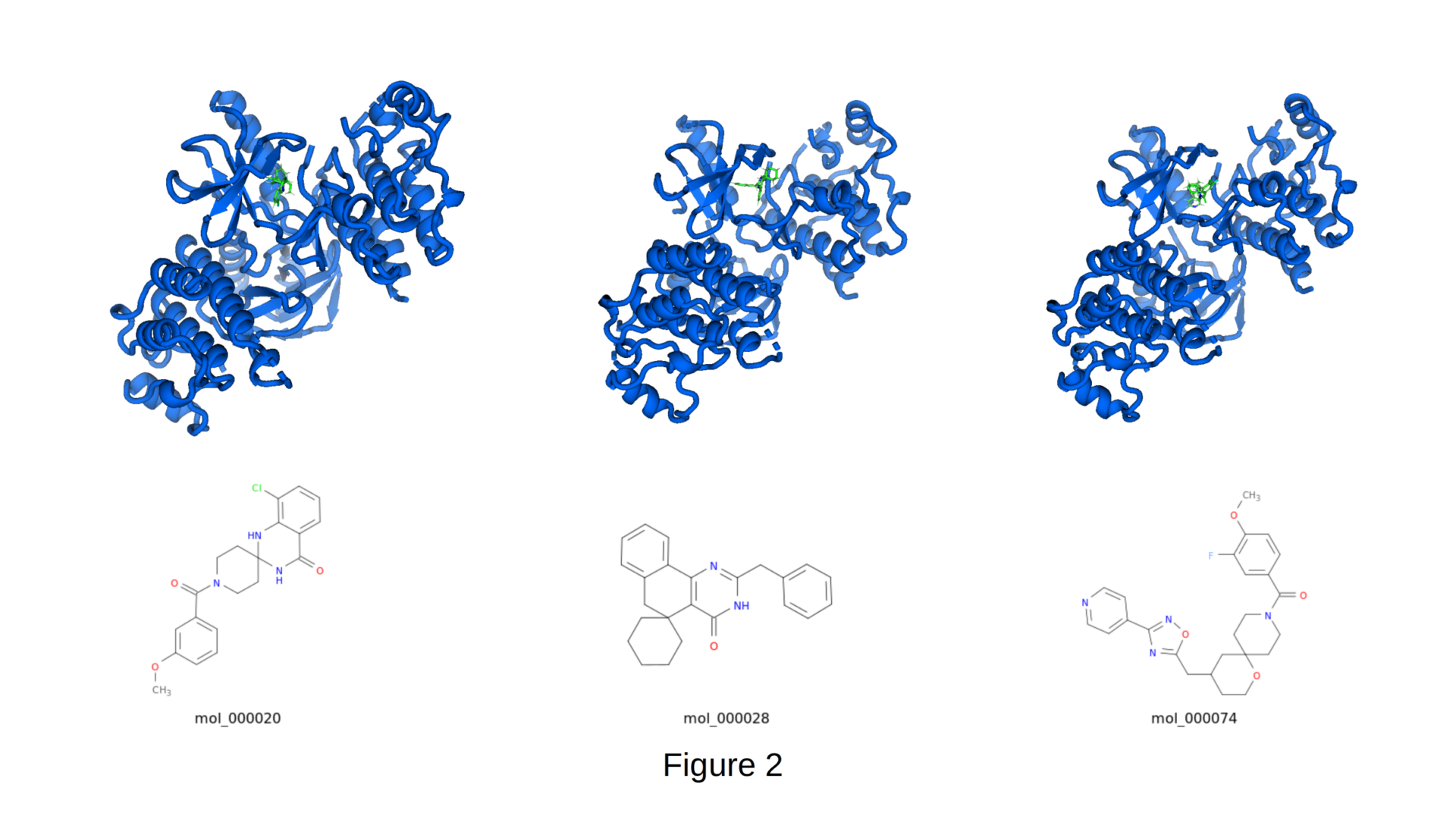
Our CIPHER-SKI platform produced a series of structurally novel, scaffold-hopped compounds that achieved strong predicted binding affinities against 4XV2, with binding scores routinely reaching -9.0 to -11.4 kcal/mol. The three representative molecules shown in Figure 2 (mol_000020, mol_000028, and mol_000074) illustrate the diversity of cores generated, including spirocyclic, fused heterocyclic, and macrocyclic-like architectures, while still forming key hinge hydrogen bonds and occupying the hydrophobic back pocket. Because our system is physics-informed (incorporating both docking scores and quantum-mechanical energies derived during molecular dynamics), the generated ligands not only dock tightly but also maintain favorable internal energies and drug-like properties (QED 0.62 – 0.85, molecular weight 350 – 500 Da).
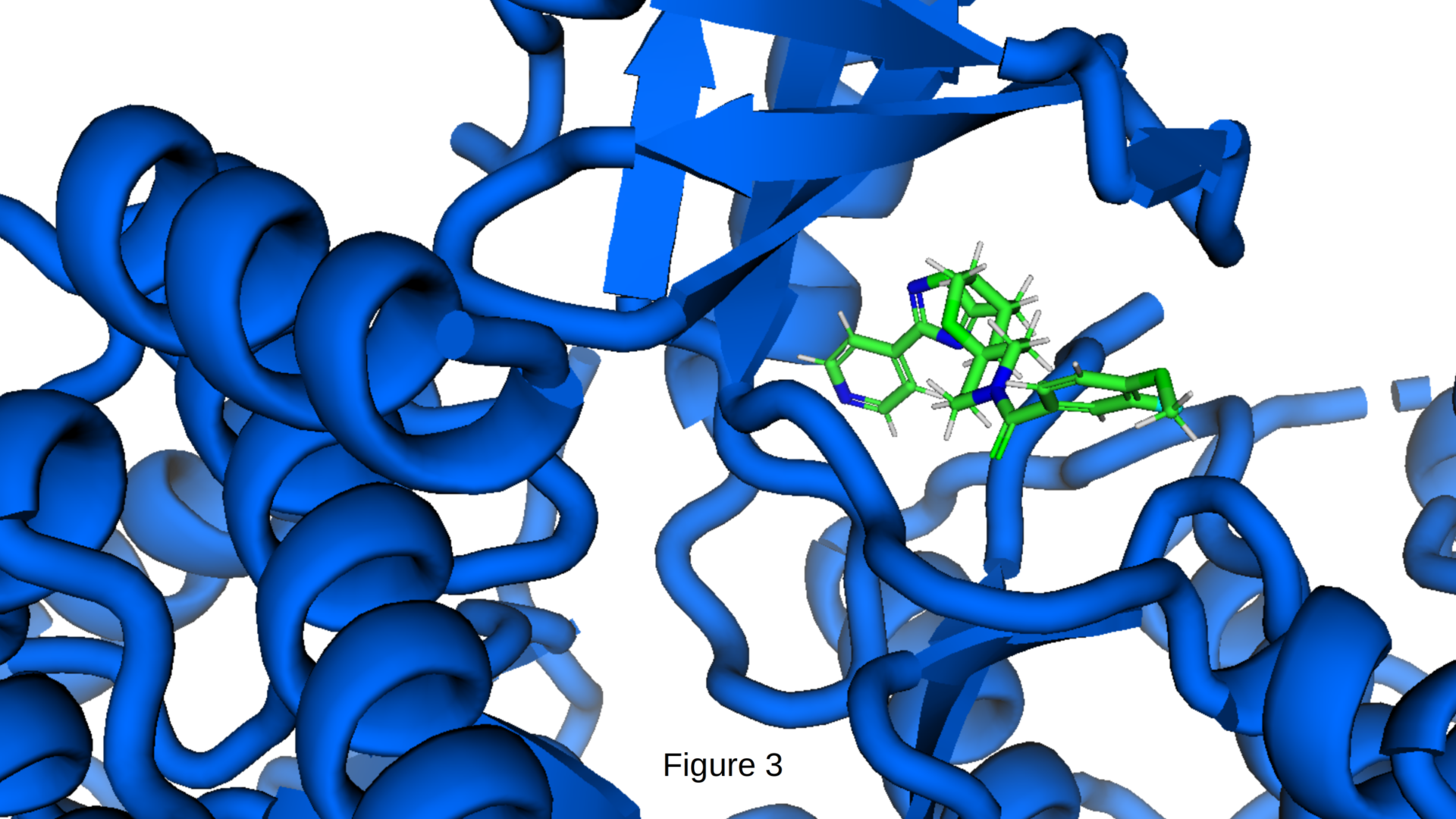
Figure 3 represents a high-resolution close-up of mol_000074 reveals the precise molecular interactions achieved through our scaffold-hopping approach. The compound adopts an extended conformation that effectively occupies the ATP-binding pocket, forming multiple key contacts. The central heterocyclic core establishes strong hinge-region hydrogen bonds (visible as blue dashed lines), while the peripheral aromatic rings engage in hydrophobic interactions with the back pocket and P-loop. The spirocyclic and ether-containing moieties provide conformational rigidity and additional polar contacts, helping the molecule maintain an energetically favorable pose.
Notably, mol_000074 achieves a strong predicted binding affinity (−10.0 kcal/mol), while retaining excellent drug-like properties. This image highlights CIPHER-SKI’s ability to generate structurally novel chemotypes that still satisfy the precise 3D pharmacophore requirements of the target, a capability that goes well beyond traditional optimization or standard generative models, which typically remain confined to known inhibitor scaffolds.
This is a significant advantage over conventional deep-learning generators, which often remain trapped within known BRAF chemotypes. By performing true scaffold hopping while optimizing for both binding affinity and energetic stability, CIPHER-SKI opens new chemical space against this high-value oncology target and may help circumvent resistance mechanisms that plague current BRAF inhibitors.
Demonstrating Power on a Challenging Target
The Switch-II pocket of KRAS G12C (PDB 6OIM), a key oncogenic driver in non-small cell lung cancer, colorectal cancer, and pancreatic cancer is considered one of the most challenging binding sites in oncology. Shallow, highly dynamic, and long regarded as “undruggable” by non-covalent molecules, it has resisted high-affinity engagement for decades. Despite these obstacles, CIPHER-SKI has generated multiple structurally novel scaffolds with strong predicted binding, as shown in Figure 4.
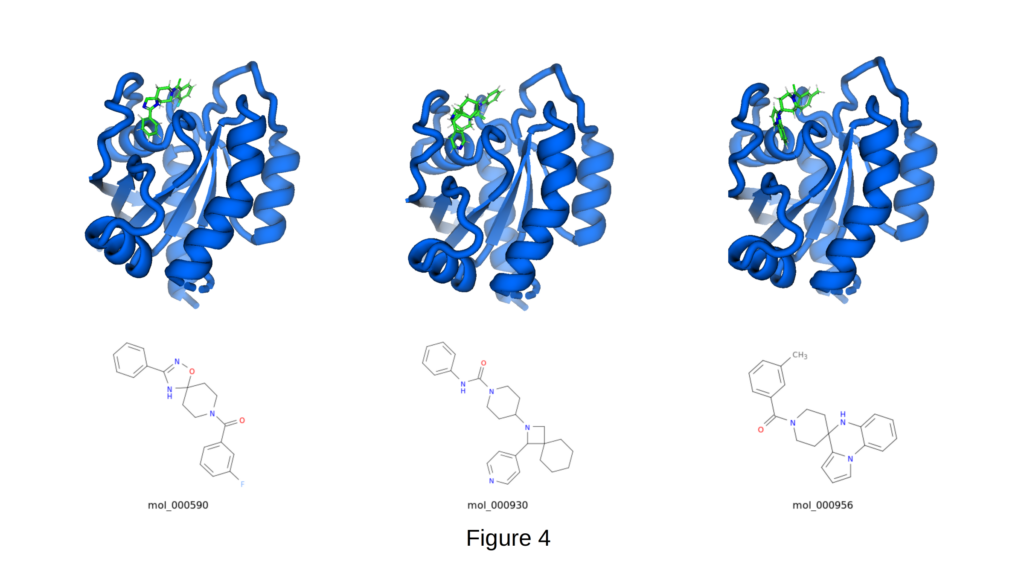
The three highlighted compounds; mol_000590 (-9.7 kcal/mol, QED 0.915), mol_000930 (-9.4 kcal/mol), and mol_000956 (-9.3 kcal/mol) adopt distinct binding modes within the same pocket while maintaining excellent drug-like properties.
Notable binding features include deep penetration into the hydrophobic core of the Switch-II region, formation of key hydrogen bonds with backbone residues, and effective filling of adjacent sub-pockets through aromatic and aliphatic groups. mol_000590, in particular, displays outstanding shape complementarity and a highly favorable binding pose.
Exceptional Binding of mol_000955 in the KRAS G12C Switch-II Pocket (PDB 6OIM) can be seen in Figure 5. CIPHER-SKI continues to deliver outstanding results on this challenging target. mol_000598 achieves a remarkable predicted binding affinity of −10.5 kcal/mol while maintaining a favorable drug-like profile (QED 0.70, LogP 4.21, SA Score 7.54, 31 heavy atoms).
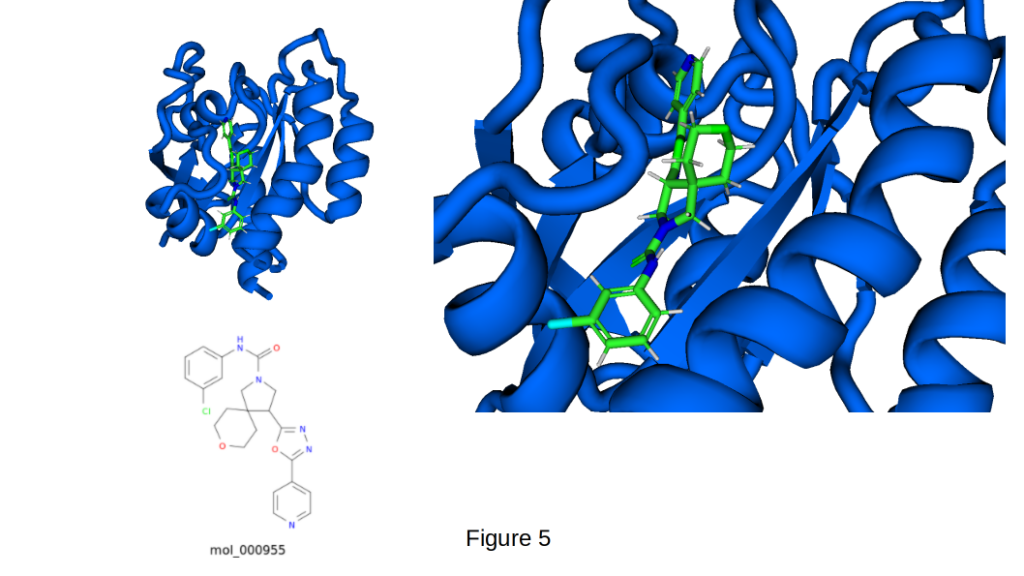
The ligand adopts an extended conformation that deeply occupies the Switch-II pocket. In the detailed view (right panel), several key interactions are visible:
- The indole and aromatic systems engage in extensive hydrophobic and π-stacking interactions with residues such as Phe78, Met72, Leu79, and Ile100.
- The polar amide and nitrogen-containing groups form multiple hydrogen bonds and polar contacts, particularly with the backbone of the Switch-II loop (e.g., near Glu62/Glu63 and Asp69).
- The molecule’s cyclopentyl and flexible linker regions effectively fill adjacent hydrophobic sub-pockets, enhancing shape complementarity.
- Overall, the ligand bridges the core hydrophobic groove and the more polar entrance of the pocket, creating a highly complementary binding pose.
With 32 heavy atoms, this compound sits in an ideal size range, large enough for high affinity yet compact enough for excellent drug-like properties. This result represents one of CIPHER-SKI’s strongest non-covalent binders and further demonstrates the platform’s ability to generate potent, structurally novel scaffolds against a historically difficult binding site.
Growing High-Affinity EphB1 Ligands Directly in the Protein Binding Pocket
CIPHER-SKI successfully generated multiple high-quality ligands targeting EphB1 by growing complete drug-like molecules directly inside the protein’s binding cavity.
Using our proprietary two-phase workflow, the system first performed intelligent scaffold hopping to identify optimal molecular cores, followed by targeted branch optimization to refine peripheral groups for enhanced binding affinity, shape complementarity, and drug-like properties.
Figure 6 shows three standout examples:
- mol_000101: -10.6 kcal/mol binding affinity
- mol_000050: -10.3 kcal/mol binding affinity
- mol_000636: -9.9 kcal/mol binding affinity
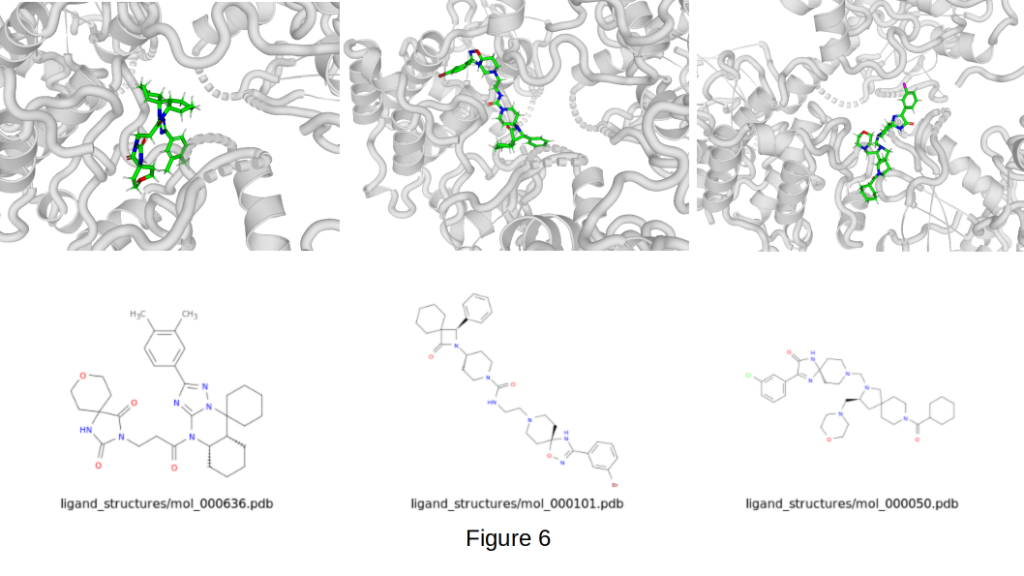
These molecules were comprehensively optimized not only for binding affinity but also for key drug-like properties, including QED, Fsp3, TPSA, rotatable bonds, H-bond donors/acceptors, molecular weight, logP, synthetic accessibility, and overall topology.
Why EphB1 (PDB 3ZFX) Matters: EphB1 is overexpressed in several aggressive cancers, including medulloblastoma, lung adenocarcinoma, colorectal cancer, and bone metastasis. It plays a key role in tumor cell proliferation, migration, invasion, and therapy resistance. Despite its clinical relevance, there are currently very few selective small-molecule inhibitors available for EphB1, representing a significant unmet medical need.
CIPHER-SKI’s ability to rapidly generate potent, multi-property-optimized, and patentable molecules for challenging targets like EphB1 highlights the platform’s power in accelerating early-stage drug discovery.
Textbook PROTAC poses generated by CIPHER-SKI on the EphB1 receptor (PDB 3ZFX)
Proteolysis Targeting Chimeras (PROTACs) are transforming modern medicine by moving beyond traditional inhibition. Instead of simply blocking a protein’s function, PROTACs recruit the cell’s own ubiquitin-proteasome system to completely degrade disease-causing proteins. This catalytic, event-driven mechanism allows a single PROTAC molecule to eliminate multiple copies of its target, offering superior potency, the ability to tackle “undruggable” proteins, and the potential to overcome resistance that defeats conventional small-molecule inhibitors.
However, designing effective PROTACs is extraordinarily challenging. It requires precise spatial positioning of the warhead and E3-binding moiety through an optimized linker. The warhead must bind deep inside the target protein pocket, the E3 ligand must be properly exposed on the outside to recruit the E3 ligase (VHL in our case), and the linker must bridge the two without clashing while enabling productive ternary complex formation (Target + PROTAC + E3 ligase). This ternary geometry, not just binary docking into the target pocket, is the real requirement for cellular degradation activity.
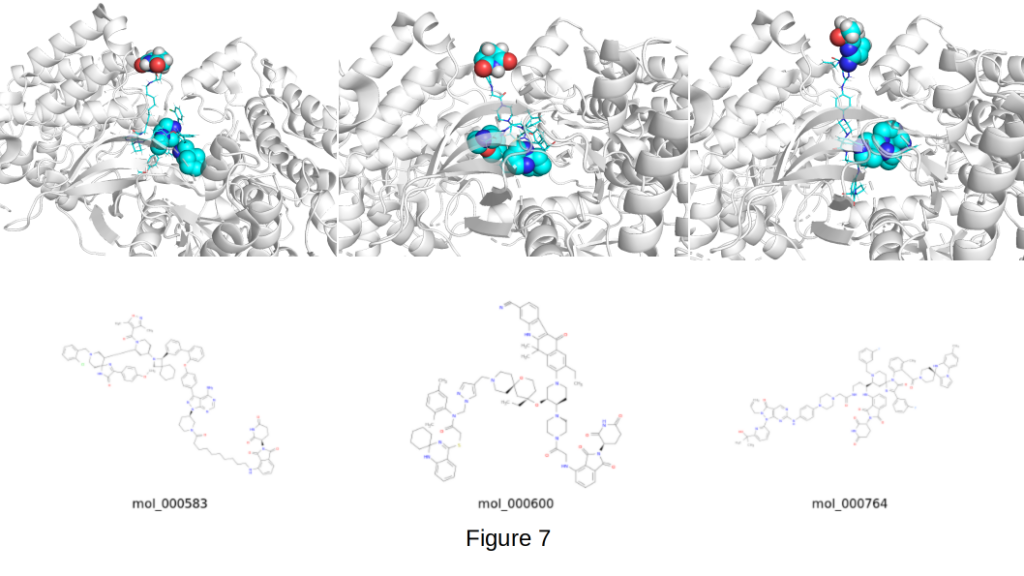
The three molecules shown above in Figure 7 represent outstanding examples of real scaffold-hopping PROTAC design:
- mol_000583: left, docking score -12.1 kcal/mol
- mol_000600: middle, docking score -11.7 kcal/mol
- mol_000764: right, docking score -11.4 kcal/mol
In all three cases the E3 ligand (shown as red/white/blue spheres) is cleanly extended outward into solvent like an antenna, perfectly positioned to recruit the E3 ligase. The two spiro centers (highlighted as blue spheres) are deeply buried in the EphB1 ATP pocket, providing the rigid, 3D warhead architecture that CIPHER-SKI was specifically designed to generate.
Particularly noteworthy is mol_000764. While it uses a slightly modified six-membered imide E3 motif (a methyl-substituted variant rather than the classic unsubstituted glutarimide), it still achieves textbook PROTAC geometry with excellent spatial separation and a fully exposed E3 end. This demonstrates that CIPHER-SKI can successfully generate high-quality, productive poses even when exploring non-standard but still validated CRBN-recruiting motifs, a valuable capability for expanding chemical space and improving drug-like properties.
Figure 8 represents a standout PROTAC pose from CIPHER-SKI: mol_001193 on the EphB1 receptor (PDB 3ZFX). Among all the molecules generated in this run, mol_001193 represents one of the strongest and most exciting results.
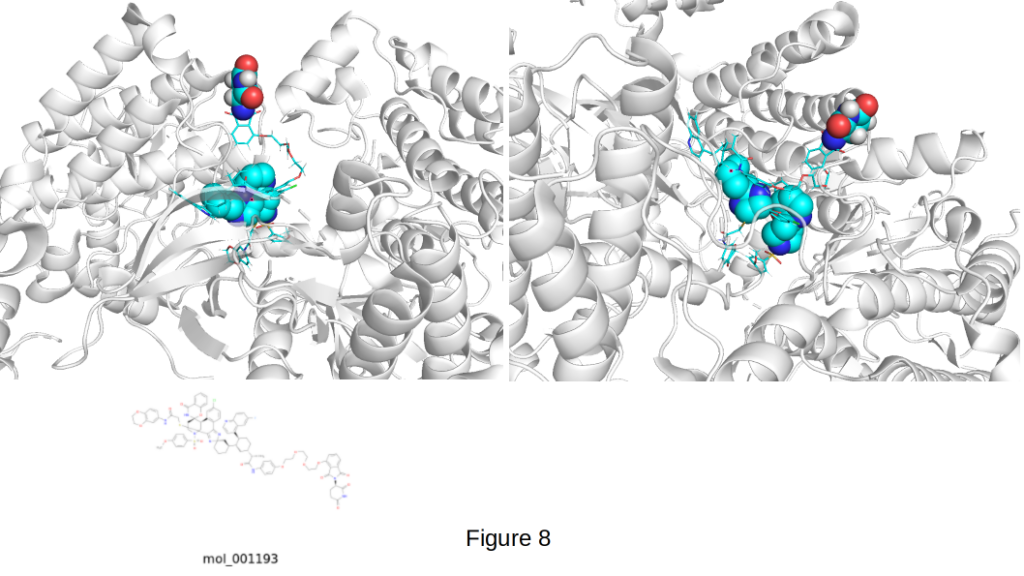
With an exceptional docking score of −13.9 kcal/mol, this molecule achieves near-perfect PROTAC geometry. The E3 ligand (shown as red/white/blue spheres) is cleanly extended outward into solvent like a well-positioned antenna, ideally placed to recruit the E3 ligase without steric hindrance. The rigid, spiro-containing warhead (cyan/blue spheres) is deeply and snugly buried in the EphB1 ATP-binding pocket, a beautiful example of real scaffold hopping. This pose is textbook in every respect: optimal spatial separation between warhead and E3 ligand, no obvious clashes, excellent complementarity with the protein surface, and clear potential for productive ternary complex formation. A docking score of -13.9 kcal/mol places mol_001193 among the very best computationally designed PROTACs we have seen to date, a result that would be considered outstanding even by experienced PROTAC medicinal chemistry teams.
This molecule (mol_001193) further validates that CIPHER-SKI is not only capable of generating novel, spiro-rich warheads with high binding affinity, but can also consistently deliver the precise 3D architecture required for functional PROTAC activity. It stands as a powerful demonstration of the platform’s ability to solve one of the hardest challenges in PROTAC design: achieving both strong target engagement and proper E3 presentation simultaneously.
These are not incremental decorations of known scaffolds. They are the direct result of real scaffold hopping: spiro-rich warheads connected through sophisticated, bridged linkers that achieve the ideal spatial separation and orientation required for productive ternary complex formation. The poses are clean, the geometry is complementary, and the E3 end is fully exposed in every case — exactly the kind of high-quality starting points that experienced PROTAC teams look for.
CIPHER-SKI is not another generative model that samples random variations of known molecules. It is a complete molecular compiler – the LLVM of drug discovery – that treats every molecule like an executable computer program. Just as a modern compiler takes source code, applies safe transformation rules, and produces faster, cleaner software, CIPHER-SKI takes simple molecular fragments and intelligently rewrites them into optimized drug candidates using the same core principles that power today’s most advanced programming compilers.
The platform specifically optimizes two critical components:
- Warhead optimization: Exploring multiple attachment chemistries and exit vectors to maximize target binding affinity while preserving synthetic accessibility.
- Linker optimization: Systematically sampling bridged and rigid spiro/PEG-like linkers of varying lengths and flexibility to achieve the ideal spatial separation and orientation required for productive ternary complex formation with the E3 ligase.
By combining these targeted optimizations with shape similarity (Ps) scoring and low-exhaustiveness local refinement, CIPHER-SKI produced the elegant, extended-linker PROTACs shown above, molecules with strong docking scores, textbook ternary geometry, and clear potential for cellular activity.
The Dual-Spiro Three-Point Hold: Scaffold Hopping + Branch Optimization
This PROTACs (mol_000064 and mol_000255) is one of the strongest examples yet of scaffold hopping + branch optimization delivered by CIPHER-SKI’s Formal Term Graph Rewriter. The rewriter applied a precise sequence of rules to transform a flexible linker into a clinically inspired architecture featuring rigid spiro centers on both the left and right sides.
The result is shown in Figure 9 which depicts a beautifully balanced three-point hold, clearly visualized in the image below with cyan spheres highlighting the key interaction regions:
- Point 1 (Warhead grip – lower left sphere): The left-side spiro-anchored warhead system locks onto the target protein with high affinity and optimal vector geometry.
- Point 2 (E3 grip – top sphere): The glutarimide on the far right recruits cereblon (CRBN) with the classic, proven binding mode, fully exposed and perfectly positioned for docking.
- Point 3 (Dual rigid anchors – lower right sphere): The two spiro centers act as powerful conformational locks, fixing distance, angle, and orientation between the warhead and E3 ligand.
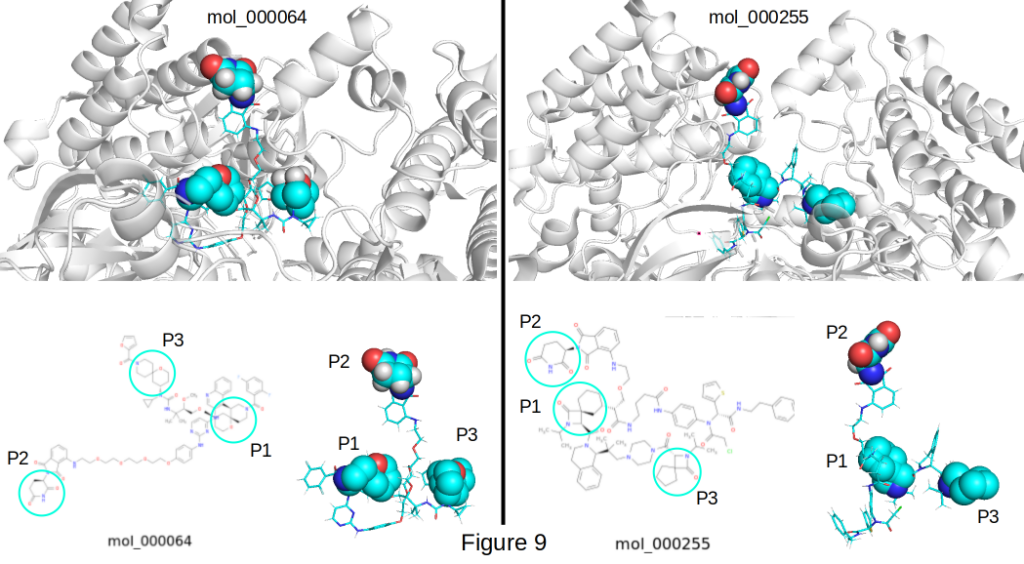
These molecules achieved excellent docking scores of -11.1 kcal/mol for mol_000255 and -10.1 kcal/mol for mol_000064 against the 3ZFX receptor model, these strong scores indicate outstanding ternary complex potential.
After the scaffold hop, the rewriter executed intelligent branch optimization, extending and rigidifying the side chains (piperazine, amide, cyclopropyl, and PEG junctions) while preserving synthetic tractability and drug-like properties. The long PEG linker is now elegantly bookended by rigid spiro anchors, giving the molecule both the flexibility needed for ternary complex formation and the rigidity required for stability.
The fully exposed glutarimide, dual-spiro architecture, and outstanding docking scores make these some of the most promising ternary complex candidates generated so far.
This is precisely what CIPHER-SKI’s PROTAC mode with Term Graph Rewriting was built to deliver: intelligent scaffold hopping followed by precise branch optimization, all in one automated, explainable step, producing molecules that look and behave like they came from a world-class medicinal chemistry team.
These results demonstrate that CIPHER-SKI is not just generating molecules, it is successfully solving one of the hardest problems in modern drug discovery: creating PROTACs with the precise 3D architecture needed for real degradation activity.
The CIPHER-SKI Advantage
We don’t just generate molecules. We compile them, intelligently rewriting molecular source code into optimized drug candidates using physics-informed scaffold kinetic intelligence.
By combining powerful learning systems with principled exploration and real energetic feedback, CIPHER-SKI navigates the vast chemical universe far more effectively than traditional methods or standard generative AI; delivering leads that are both novel and set a clear roadmap for developing a commercial drug. The result? Faster discovery. Stronger IP. Better chances of clinical success.